
While engineering, finance, and commerce have profited immensely from novel algorithms, they are not the only ones. Large-scale computation has been an integral part of the toolkit in the physical sciences for many decades - and some of the recent advances in AI have started to change how scientific discoveries are made.
There has been a lot of excitement about prominent achievements in the physical sciences, like using machine learning to render an image of a black hole or the contribution of AlphaFold towards protein folding. This article will cover some of the more prominent usages of AI in chemistry, the parent discipline of the aforementioned protein folding problem.
One of the chief goals of chemistry is to understand matter, its properties, and the transformations it can undergo. Chemistry is what we turn to when we are looking for a new superconductor, a vaccine, or any other material with the properties we desire.
Traditionally, we think of chemistry being done in a lab with test tubes, flasks, and gas burners. But it has also benefited from developments in computing and quantum mechanics, both of which rose to prominence in the early-mid 20th century. Early applications included using computers to help solve physics-based calculations; by blending theoretical chemistry with computer programming, we were able to simulate (albeit far from perfect) chemical systems. Eventually, this vein of work grew into a subfield now called computational chemistry. The subfield started to gain momentum in the 1970s and was featured in the Nobel Prizes of 1998 and 2013. Even so, while computational chemistry has gained more and more recognition over the past few decades, its importance has been largely overshadowed by that of lab experiments - the cornerstone of chemical discovery.
However, with current advancements in AI, data-centric techniques, and ever-growing amounts of data, we might be witnessing a change where computational approaches are used not just to assist lab experiments but to guide them.
The Chemical Discovery Process
So how is AI enabling this shift? One particular development is the application of machine learning to material discovery and molecular design - two core problems in chemistry.
In the traditional approach, molecules are designed in roughly four stages, as outlined in the figure below. It is important to note that each stage can take years and many resources with no guarantee of success.
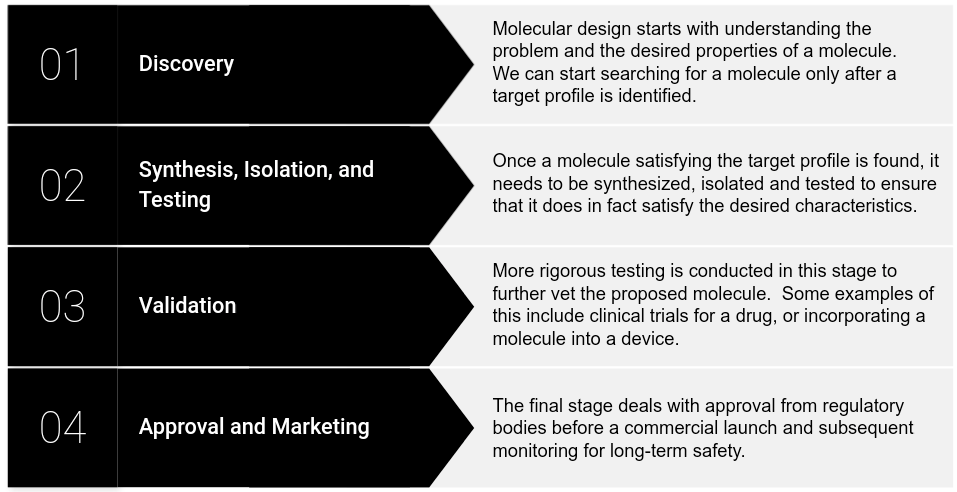
The discovery stage relies on theoretical frameworks that have been developed over centuries to guide molecular design. However, when looking for materials that are “useful” (e.g. vaseline, Teflon, penicillin), we must remember that many of them come from compounds commonly found in nature. Moreover, the utility of these compounds is often discovered after the fact. The opposite of that - targeted search - is an endeavor that would require much more time and resources (and even then, one would likely have to use known “useful” compounds as starting points). To give the reader some perspective, it has been estimated that the pharmacologically active chemical space (i.e. the number of molecules) is 1060! Manual search in such a space would take enormous time and resources, even before the testing and scaling phases.
So how does AI come into all of this, and how is it accelerating chemical discovery?
First, machine learning has improved existing methods of simulating chemical environments. We have already mentioned that computational chemistry allows us to partly bypass lab experiments. However, computational chemistry calculations simulating quantum mechanical processes scale very poorly in both computational cost and accuracy of the chemical simulation. The underlying core problem in computational chemistry is solving the electronic Schrödinger equation for complex molecules - that is, given the positions of a collection of atomic nuclei and the total number of electrons, calculate the properties of interest. An exact solution is possible only for one-electron systems, and for the rest, we must rely on “good enough” approximations. Additionally, many popular methods for approximating the Schrödinger equation scale exponentially, making a brute force solution intractable. Over the last century, many approaches have been developed to speed up the calculation without sacrificing too much accuracy; however, even some of the “cheaper” methods can cause computational bottlenecks.
One way that AI has accelerated these calculations is by blending them with machine learning. Another approach bypasses modelling the physical process altogether by directly mapping molecule representations to the desired property. Both of these approaches allowed chemists to screen chemical databases more efficiently for various properties such as atomic charges, ionization energies etc.
The Rise of Generative Chemistry
While faster calculations are an improvement, it does not address the fact that we are still limited to known compounds - which is only a fraction of the active chemical space. We still have to manually specify the molecules we want to profile. How can we reverse this paradigm and design an algorithm that will search the chemical space and find the right candidate(s) for us? The answer may lie in the application of generative models to the problem of molecular discovery.
But before we get to that, it is essential to talk about how chemical structures can be represented digitally (and which ones can be used for generative modelling). Many representations have been developed over the last few decades, most of which fall under one of the four categories below:
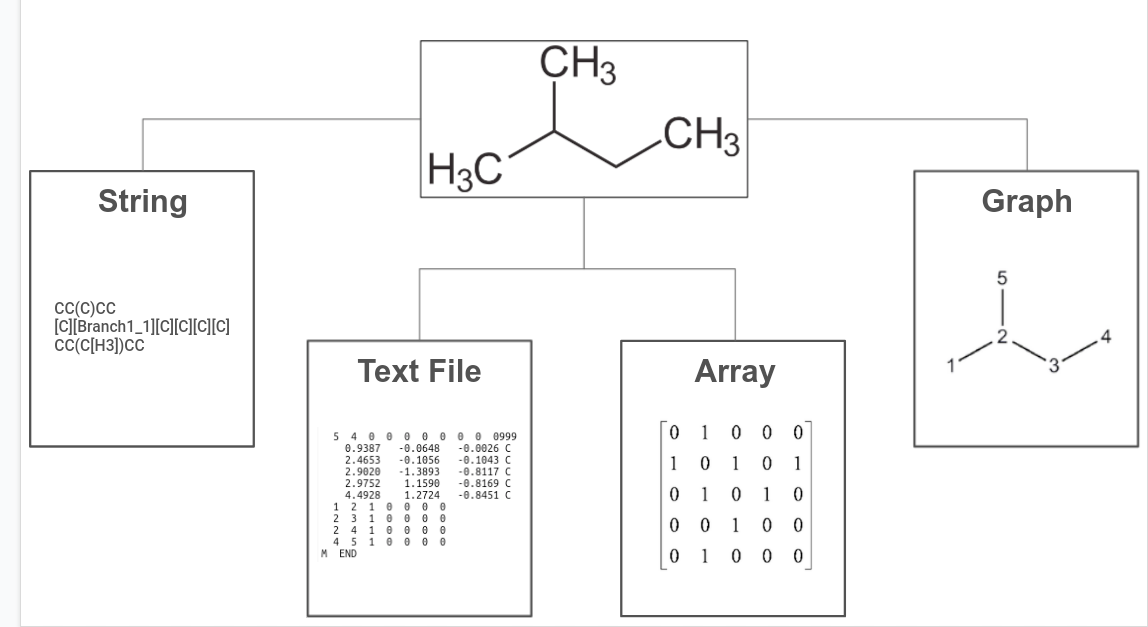
Unsurprisingly, chemical structures can be represented as arrays. Originally, array representations of molecules were used to assist in chemical database searching; in the early 2000s, however, a new type of array representation called Extended connectivity fingerprints (ECFPs) was introduced. Designed specifically to capture features relevant to molecular activity, ECFPs are often among the first representations tested when trying to predict molecular properties.
Chemical structure information could be also dumped into a text file - a common output for quantum chemistry calculation. These text files can be very rich in information, however, they are generally not very useful as inputs for machine learning models. String representations, on the other hand, have much of the information encoded in their syntax; this has made them particularly amenable to generative modelling, much like text generation.
Lastly, graph-based representations are natural approaches that allow us not only to encode atom-specific properties in node embeddings but also to capture chemical bonds in edge embeddings. Additionally, when combined with message passing, these approaches allow us to account for (and configure) the influence on nodes from its neighbours, which mirrors how atoms in a chemical structure influence each other. These properties make graph-based representations a go-to type of input representation for deep learning models.
The types of representations above can have their own subtypes; it is also, unfortunately, inconclusive which type of representation works best for any specific problem. For example, array representations are usually preferred for property prediction but graph representations have also become a strong contender in the last couple of years. It is also important to note that depending on the problem, multiple types of representation can be used in conjunction with each other.
So how (and which?) representations can be used to explore the chemical space? We have already mentioned that string representations are amenable to generative modelling. Graph representations were less susceptible to generative modelling at first but have recently become a strong contender thanks to variational autoencoders (VAEs); the latter have proven especially useful as they allow us to have a continuous, more machine-readable representation. One study used VAEs to show that both strings and graphs can be encoded and decoded into a latent space, where molecules are no longer discrete but rather real-valued continuous vectors that could be decoded back to discrete molecular representations (which may or may not be valid); Euclidean distance between different vectors would correspond to chemical similarity. Another model is added between the encoder and decoder to predict target properties from any point in the latent space.
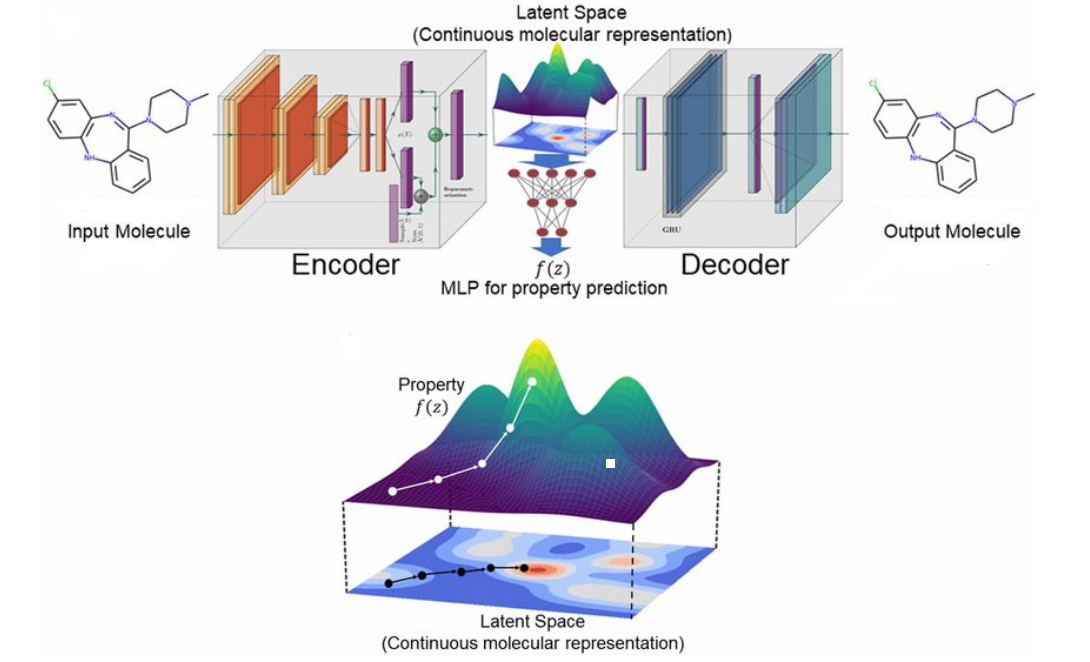
But while generating molecules in and of itself is an easy task - one can just take any generative model and apply it to the representation they want - it is a much more challenging problem to generate structures that are both chemically valid and exhibit the properties we want.
Initial approaches to achieving this involved pretraining models on existing datasets, which could then be used for transfer learning. Biasing a model via a calibration dataset allows generating structures skewed towards particular properties, which could be further calibrated using a different algorithm, such as reinforcement learning. Several examples of this involved using string or graph representations; however, they struggled with chemical validity or were not successful enough at getting to properties desired. Additionally, relying on a pretraining dataset limits the search space and introduces perhaps unwanted bias.
One attempt at breaking free from pretraining uses a Markov decision process (MDP) to ensure the validity of chemical structures and optimizes the MDP for desired properties with Deep Q-Learning. One particular advantage of this model is that it allows the user to visualize the favourability of different actions. The figure below shows how this idea works in practice; incremental steps deemed advantageous by the model are made from a starting structure to maximize a particular property.
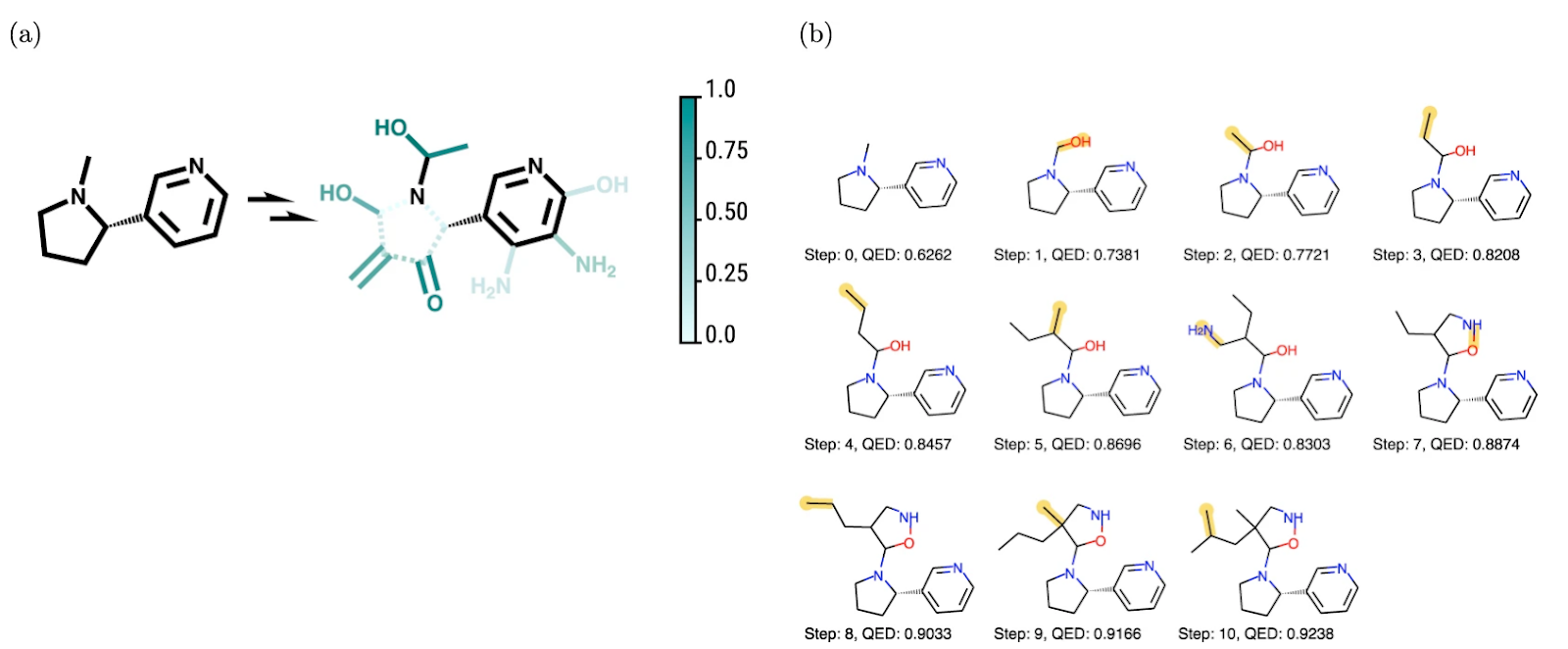
Despite being in its infancy, the use of AI to explore chemical space has already shown some great promise. It has given us a new paradigm for exploring the chemical space; a new way to test theories and hypotheses. And while not as accurate or grounded in empiricism as experimental research, computational approaches will remain an active area of research for the foreseeable future and are already part of any research team.
Other Use Cases and Challenges Ahead
So far, we have talked about how AI is helping discover new chemicals faster by leveraging generative algorithms for searching the chemical space. While it is one of the more notable use cases, it is by no means exhaustive. AI is being applied to many other problems in chemistry, some of which include:
- Automating lab work. We can use machine learning techniques to accelerate the synthesis workflow itself. One approach uses “self-driving laboratories” to automate mundane tasks, optimize resource spending, and save time. While still relatively new, one notable case is the use of the robotic platform Ada to automate the synthesis, processing, and characterization of thin-film materials (see the platform in action here). Another study showcased the use of a mobile robotic chemist capable of operating instruments and performing measurements for 688 experiments over eight days.
- Reaction prediction. We can use a classification model to predict the type of reaction that will occur, or simplify the problem and predict whether or not the reaction will occur. There are many different approaches to this modelling problem.
- Chemical data mining. Like so many other disciplines, chemistry has an enormous body of scientific literature that can be used for studying trends and correlations. One notable example is the use of data mining on the vast quantity of information supplied by the Human Genome Project to identify trends in genomics data.
Finally, while the new data-driven trends are evolving rapidly and are already making a difference, they also present us with many new challenges, including:
- The gap between computation and experiment. While the goal of computational approaches is to help achieve the goals of an experiment, the results from the former are not always transferable to the latter. For example, when looking for molecule candidates using machine learning, one has to keep in mind that molecules are rarely unique in their synthetic pathways, and it is often difficult to know whether an unexplored chemical reaction will work in practice. And even if it will, there are issues concerning yield, purity, and isolation of the target chemical compound. The gap between computational and experimental work is made even wider due to metrics employed in the former not being always transferable to the latter (the QED mentioned above is just one example of many), and that experimental validation may not be feasible.
- The need for better databases and absence of benchmarks. The overall chemical space is infinite, so the most we can hope for is a large enough sample that will help us generalize. However, most current databases were designed for different purposes, often using different file formats; some of them lack validation procedures for making submissions, or they were not designed with AI tasks in mind. Additionally, most databases we have are limited in their chemical scope - they only contain certain types of molecules. Finally, most tasks involving chemical predictions using AI lack benchmarks, making the comparison of many different studies unfeasible.
One of the chief reasons for AlphaFold’s success is that it had all of the above supplied as part of the Critical Assessment of protein Structure Prediction (CASP) competition, showing that an organized effort is required to streamline and improve other tasks involving chemical predictions.
Summary
As we continue heading into the digital age, new algorithms and more powerful hardware will continue lifting the veil behind previously intractable problems. The integration of AI into chemical discovery is still in its infancy - but it is already not unusual to hear the term “data-driven discovery.” Numerous companies - be it pharmaceutical giants or younger startups - have already adopted many of the techniques mentioned above, and with them, brought increased automation, efficiency, and reproducibility to chemistry. AI is allowing us to conduct science on unprecedented scales, and over the last few years, this has engendered numerous initiatives and attracted funding that will continue leading us further into the age of autonomous scientific discovery.
Writer Profile
Renée Gil is a PhD student at Carleton University, advised by Prof. Christopher Rowley. Her research is in the field of cheminformatics, particularly the use of machine learning for studying covalent reactivity between molecules and proteins.
Citation
For attribution in academic contexts or books, please cite this work as
Renée Gil, "How AI is Changing Chemical Discovery", The Gradient, 2022.
BibTeX citation:
@article{canogil2022chemistry,
author = {Renée, Gil},
title = {How AI is Changing Chemical Discovery},
journal = {The Gradient},
year = {2022},
howpublished = {\url{https://thegradient.pub/how-ai-is-changing-chemical-discovery} },
}


{kind=link}
{kind=link}
{kind=link}
{kind=link}